1. Molecular Oncology - R2 Practical - Colorectal Cancer¶
Use bioinformatics tools to perform data analysis on colorectal cancer omics data sets
1.1. Introduction¶
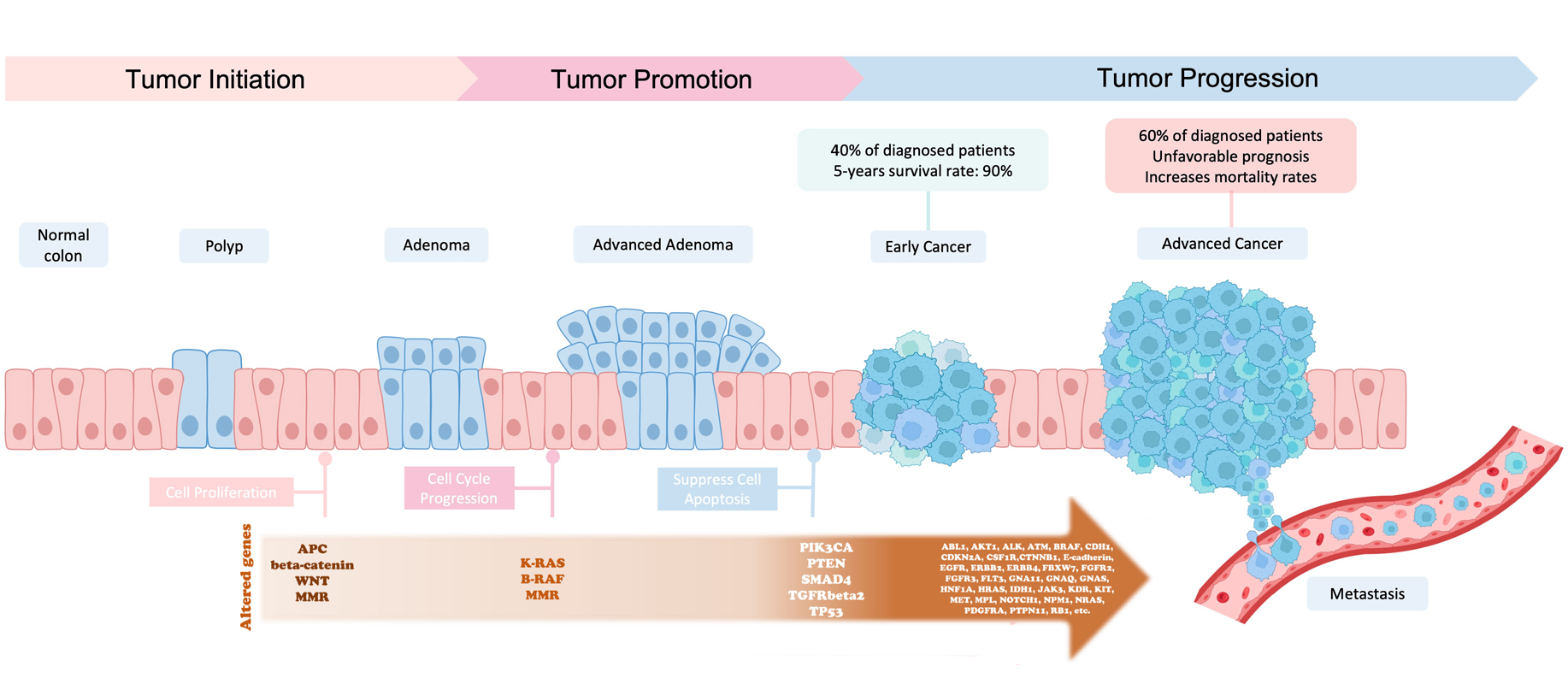
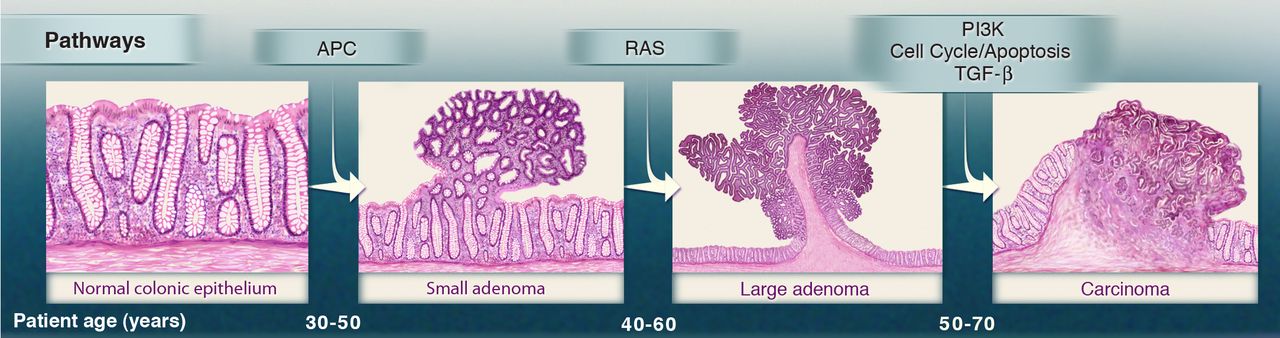
In the late 1980s, the Vogelstein model was proposed. It introduced the concept of a stepwise accumulation of genetic mutations leading to the development of colorectal cancer (CRC).
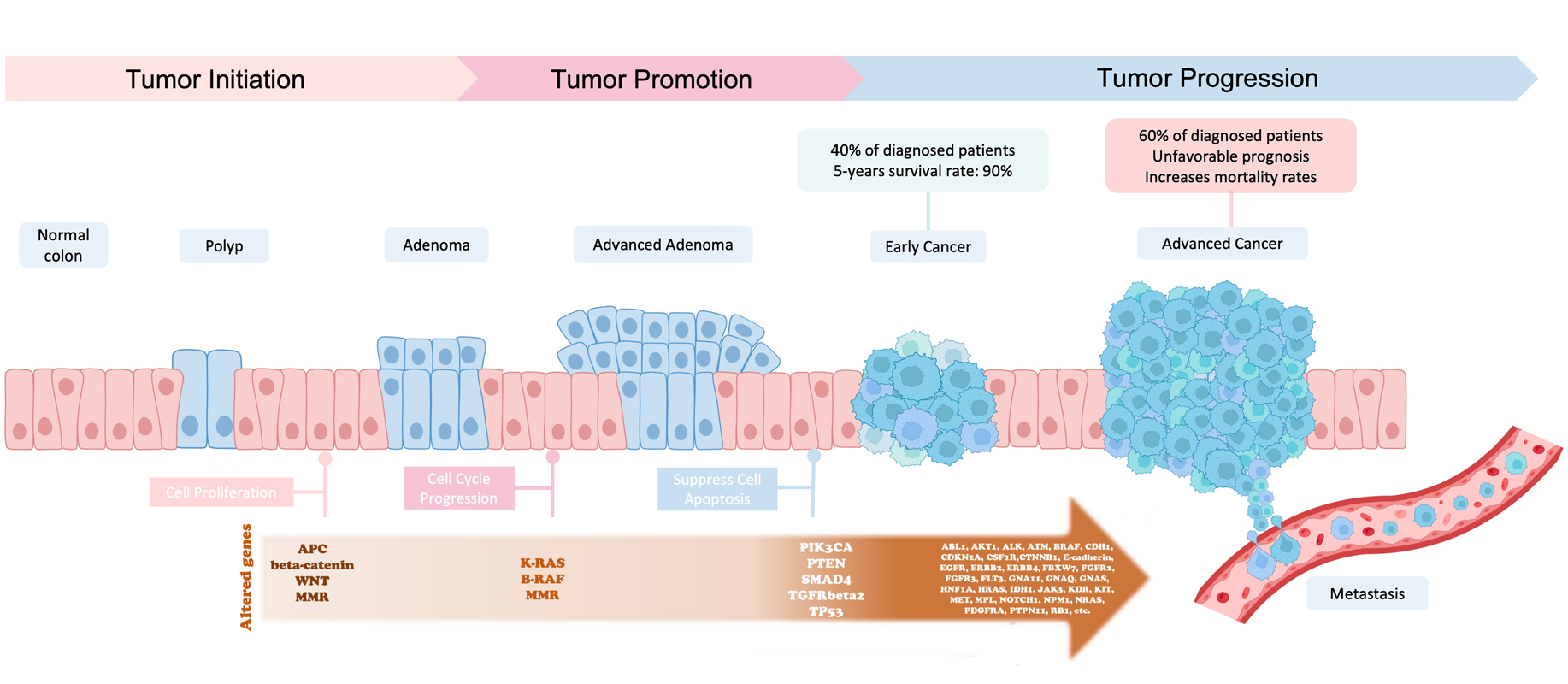
Accumulation of mutations advancing colorectal cancer development
(Figure sources: https://doi.org/10.3390/ijms241311023, https://doi.org/10.1016/j. tranon.2021.101131)
Similar to the picture above, the model highlights the importance of key genetic mutations during CRC progression,
including mutations in APC, KRAS, and TP53. While the Vogelstein model has provided a valuable foundation for
understanding colorectal cancer, subsequent research has revealed that the disease is more complex and heterogeneous
than initially described. Colorectal cancer can involve various genetic and epigenetic changes. Additional
factors, such as the tumor microenvironment, inflammation, and the immune system, also play significant roles in the
progression of the disease.
Colorectal cancer is the third most common cancer worldwide, according to the World Health Organization, accounting
for approximately 10% of all cancer cases, and it is the second leading cause of cancer-related deaths worldwide.
Research is needed to understand the mechanisms underlying treatment resistance and to develop strategies to
overcome it. Better identification and characterization of multiple CRC subtypes could guide treatment decisions and
improve the outcomes for individuals with colorectal cancer. Furthermore, markers for early detection and prevention
might allow for interventions before advanced mutations occur. Clearly it is crucial to understand the diversity and
complexity of colorectal cancer in order to develop new and effective targeted treatment strategies.
Bioinformatics tools enable the analysis of vast amounts of omic and clinical data, helping researchers identify
genetic mutations, epigenetic aberrations, biomarkers, and potential therapeutic targets in order to better understand
and combat cancer.
Web-based genomics analysis and visualization platform R2(Figure source: https://r2.amc.nl)
Today you will use advanced bioinformatics tools to explore, analyze and visualize colorectal cancer data in search for a deeper understanding. You will use the freely available and web-based genomics analysis and visualization platform R2, that is developed at Amsterdam UMC and widely adopted by researchers worldwide. R2 provides the user with many experimental and clinical data sets coupled to a wide variety of clickable bioinformatics tools. Without any coding you will gain hands-on research experience with colorectal cancer omics data and bioinformatics tools.
The in this document will open up a Google form, one per section, with which you can submit answers.
1.2. Normal colonic epithelium vs adenomatous tissue: a first impression of genomic data¶
Colorectal cancers are believed to arise predominantly from adenomas. A fundamental query in cancer research consistently revolves around understanding the distinctions between the transcriptomic profiles of normal tissue and tumor tissues. Let’s get acquainted with R2 and its large collection of omic datasets while immediately exploring differences in gene expressions between normal colonic mucosa and colorectal adenomatous tissue.
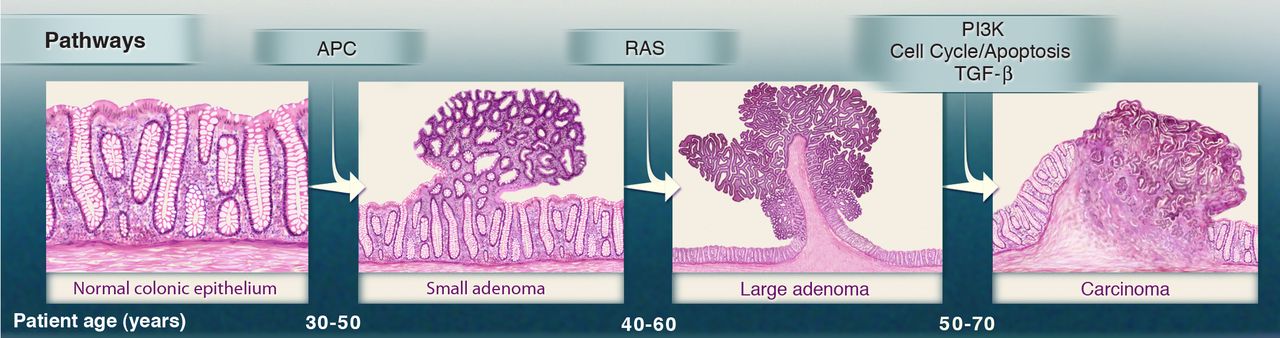
Normal tissue, precancerous adenomas and cancer growth(Figure source: https://doi.org/10.1126/science.1235122)
Datasets used:
- Mixed Colon - Marra - 64 - MAS5.0 - u133p2
1.2.1. Filtering and exploring¶
- Open a Chrome browser and use your R2 account to sign in in the collaborator’s server of the R2 platform: https://hgserver2.amc.nl (also accessible via https://r2platform.com/hg2)
Generally speaking, the R2 platform is easily accessible by the links https://r2.amc.nl | r2platform.com, but today we work from our collaborator’s server hgserver2.
You’re now on the R2 main page. This genomics analysis and visualization platform contains a wealth of data and
bioinformatics tools to analyze the datasets. Step by step, researchers are guided through a web of options
for data analysis with mostly clickable items. R2’s main page shows this principle: follow the numbered
boxes to develop your analysis of
choice.
Let’s follow these steps to get a first look at gene expressions in one of the colon cancer datasets that is hosted in R2, a
dataset that is called Mixed Colon - Marra - 64 - MAS5.0 - u133p2.
Datasets have a structured naming in R2, using the following rules:
Category - Tissue/ Tumor - author - number of samples (N) - normalization - chiptype.
In our case the dataset name tells us that the dataset contains
normal and tumor samples (mixed) of colon tissue; Marra is the author and there are 64 samples.
- We leave Box 1 as is, because we will look only at a single dataset.
- In order to find the above-mentioned dataset in R2, click on the text of the currently selected dataset in box 2.
A grid pops up that shows all the datasets that are currently available to you. Each row is a dataset and
each column contains a different searchable characteristic of the datasets.
In the bottom right corner of the grid, you can find the number of rows, i.e. available datasets.
- Under the header Tissue/Tumor type the keyword colon in the white text-field filter, and check the adapted number of rows in the bottom right corner to find out how many data sets R2 is hosting with the keyword colon in its name.
- Find the RNA expression dataset from Author Marra (thus type Marra in the Author column search field) and click on the row of the dataset that contains 64 samples (N).
- In the information panel below the grid, you find more information about this dataset. Quickly glance over the summary of the study.
- Select the dataset with a click on the blue button Confirm selection. Check on the main page in box 2 that the correct set has been selected.
Of course, it is nice to have a lot of RNA expression datasets to analyze and explore, but without proper sample annotation you have very limited analysis options. Let’s explore the annotation for the Marra dataset.
- In box 3, select the analysis type Cohort Overview and click Next.
- In the grid, you can see all the samples in rows, with the available annotation in the columns. Scroll quickly down the grid to take a glance at the possible values for each annotation.
In R2, samples of a dataset can be annotated with e.g. clinical data or biological information. Each group of annotated data is called a Track in R2. These tracks can be used to filter, color or split data in all types of R2 analyses.
Above the grid, you can find an interactive pie chart for each track. The pie charts present an overview of the track values in the dataset, detailing the percentage of samples in each category. You can hover over the tracks and click on a section to filter the grid underneath for samples with that track value.
- Check the different group values in the pie chart of the tissue track and the proportion of samples for each group.
The button below brings you to the form in which you can submit your answers for the first section.
![]()
In the dataset information panel you could read in the summary of the Marra dataset:
“Colorectal cancers are believed to arise predominantly from adenomas. Although these precancerous lesions have been subjected to extensive clinical, pathological, and molecular analyses, little is currently known about the global gene expression changes accompanying their formation. To characterize the molecular processes underlying the transformation of normal colonic epithelium, we compared the transcriptomes of 32 prospectively collected adenomas with those of normal mucosa from the same individuals.”
We had a look at the track tissue above. This track annotates which samples originate from normal mucosa and which were obtained from adenomas.
- Can you think of specific genes or pathways that you expect to show a difference in expression between the groups?
1.2.2. Find different expression profiles between normal and adenoma tissue¶
As you have seen above, R2 provides access to thousands of datasets. All these datasets can be readily analyzed with a comprehensive range of bioinformatics tools without coding. One of these tools is the “Find differential expression between groups”. The differential expression analysis aims to identify genes that are significantly different between two groups of a dataset.
- Click on Main in the upper left corner.
- Check if you have selected the Marra dataset with 64 samples in box 2 and in box 3 select type of analysis, Differential expression between two groups. Click Next.
- R2 offers a couple of statistical Test options, in this case we use the T-test which is selected by default.
- Now we have to select which grouping variable to use. Select Group by Tissue (2cat) to use the previously seen tissue annotation. And click Next.
- An extra field of settings is shown. Select Group 1 normal and Group 2 adenoma. Check that the default Transformation Log2 is selected, and P-value cutoff 0.01. Click Submit.
R2 has generated a large list of differentially expressed genes. On the right hand side of the page you find buttons to follow-up analyses, and underneath the buttons are informative tables about the genes list. One table shows how many genes have higher expression in adenomas compared to healthy tissue and vice versa.
![]() How many genes were significantly upregulated in adenomas and how many were
downregulated?
How many genes were significantly upregulated in adenomas and how many were
downregulated?
Next to the many publicly available datasets, R2 also hosts many curated lists of genes that we call
gene sets. These gene sets can be used to restrict or filter an analysis as well.
We can adapt our current search by scrolling down to the end of the page, where you find the Adjustable Settings menu. You can now use a Gene set to
restrict your list of differentially expressed genes between normal tissue and adenomas that is specifically associated with colorectal cancer.
- Click on Select gene set and a grid box will pop-up.
- Use the search field on the top of the table and fill in colorectal, hit enter.
- The KEGG (Kyoto Encyclopedia of Genes and Genomes) database is a comprehensive bioinformatics resource that integrates information about genes, proteins, pathways, and diseases. Click on the triangles in front of KEGG pathways and its subcollections, until you find the Colorectal_cancer (62) gene set. Check the checkbox in front of the gene set and hit the green Confirm selection button.
- Always click Submit after you have changed something in the Adjustable settings menu in order for the changes to take effect.
- Check out the list and see if you recognize multiple genes. You can hover over the magnifying glasses in front of each row to learn more about the genes.
- Now click on the magnifying glass in front of AXIN2 to obtain a violin plot with the scatter points of the expression value of this gene
for each sample in the dataset.
The plot shows the two groups of the tissue track in violin plots with the individual sample values showing as scatter on top. If you hover your mouse over any of the violins, you can see the group statistics. Also note you can hover over the dots in the graph to get more information of the individual samples. - The green bar in the top allows you to easily go to the next or previous gene of your list. Click on the arrow with MYC on the right side of the green bar to view this gene’s expression in the samples.
![]() We evaluate differential gene expression using both the p-value and the log fold change. What does each of these metrics tell us about the differences in gene expression?
We evaluate differential gene expression using both the p-value and the log fold change. What does each of these metrics tell us about the differences in gene expression?
What did you observe about the gene expression of AXIN2 and MYC? When you think about biological processes, why would this be?
1.2.3. Heatmaps and the WNT pathway¶
The WNT pathway is an important signal transduction cascade that plays an important role in diverse biological processes. The dysregulation of the WNT pathway has been observed in several cancers including colon cancer.
In the next sections we will regularly be using heatmaps to find subgroups of samples or genes that show similar expression profiles. Heatmaps perform unsupervised hierarchical clustering of samples. The algorithm uses the distribution of the (expression) data to find clusters that have similar (expression) profiles and shows the clusters of samples in the plot based on their (dis)similarity. This is combined with the clustering of the genes based on their expression throughout the samples. The heatmap is colored by the z-scores of the samples’ gene expression values. Often annotation tracks are shown above a heatmap. Remember that we can see this annotation but that the heatmap algorithm did not use this information to look for subgroups in the data, it uses expression values only.
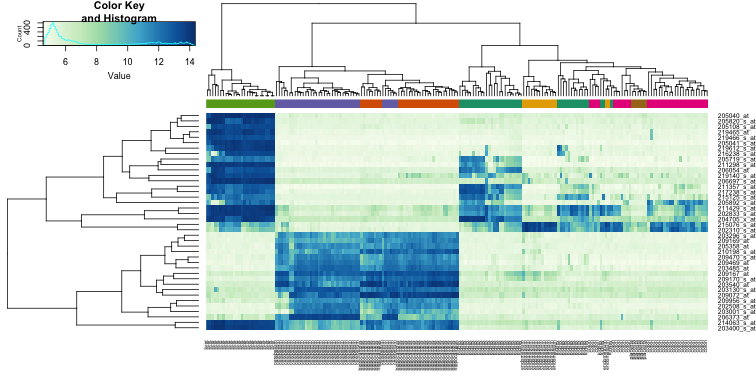
Example heatmap: finding subgroups in your data
- Go back to the gene list result page of the previous Differential Expression between two groups analysis, the tab should still be open.
- Generate a list of genes which are differentially expressed comparing normal and adenoma tissue within the KEGG WNT
pathway by adjusting the settings if needed:
- Use the False Discovery Rate for multiple testing correction,
- log2 values
- and P <0.01.
- Click on the currently selected gene set, Clear selection with the red button and find the KEGG Wnt pathway geneset (hint: use keyword Wnt). Don’t forget to hit Submit in order to redo the analysis with the new settings.
- Use the grey Heatmap(zscore) button on the right side of the page, which creates a heatmap from the gene list you just generated.
![]() Inspect the heatmap. Did you expect this pronounced clustering?
Inspect the heatmap. Did you expect this pronounced clustering?
Now let’s generate a Wnt pathway heatmap from a different route:
- Go to the main page.
- Select the analysis View Geneset (Heatmap)
- On the next page, click on Select gene set to find the KEGG Wnt_signaling_pathway. Click Submit.
![]() What does the annotation above the heatmap tell you?
What does the annotation above the heatmap tell you?
![]() How and why does this heatmap differ from the previous Wnt pathway heatmap?
How and why does this heatmap differ from the previous Wnt pathway heatmap?
(Hint: Think about the criteria used to include genes in each heatmap)
1.3. Identifying groups and their characteristics: CMS¶
Let us now move past the precancerous stage of adenomas and look at colorectal cancer. Colorectal cancer is a complex and heterogeneous disease, characterized by a multitude of variations in its genetic, molecular, and clinical attributes. This heterogeneity manifests in diverse ways, influencing the tumor’s behavior, response to treatments, and patient outcomes. Understanding this heterogeneity is critical for tailoring effective therapies and improving patient care. In this context, we will explore the various dimensions of heterogeneity in colorectal cancer and its implications for diagnosis, treatment, and research.
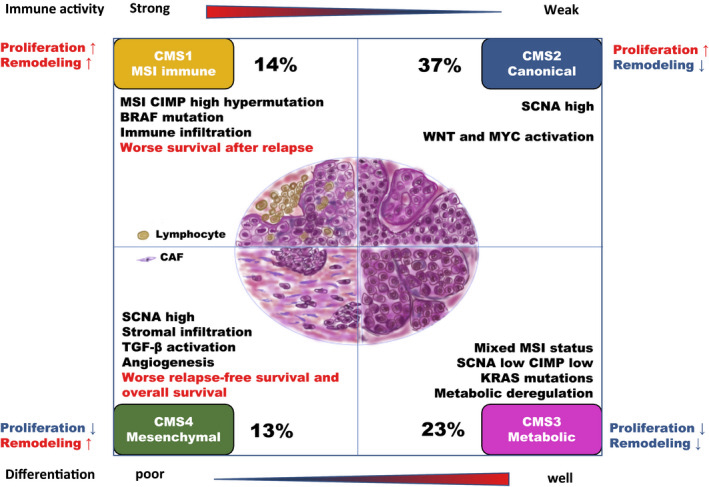
In 2015, Guinney et al. (Nat Med. 2015 Nov; 21(11):1350–1356) published a bioinformatics study on a vast collection of colorectal cancer cohorts with detailed molecular annotation. The consortium developed a now widely accepted molecular classification system that allows researchers to categorize most colorectal tumors into one of four distinct and robust subtypes, each characterized by its unique biological features. These subtypes are: CMS1 (MSI Immune), CMS2 (Canonical), CMS3 (Metabolic), and CMS4 (Mesenchymal), see the figure below.
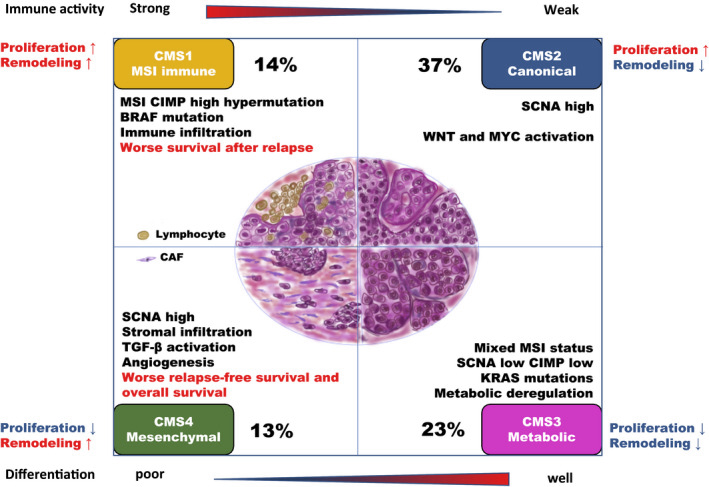 Subtypes in colorectal cancer: CMS classification
Subtypes in colorectal cancer: CMS classification
(source: http://dx.doi.org/10.1002/ags3.12362)
The button below brings you to the form in which you can submit your answers for the next section.
1.3.1. Clustering with t-SNE maps¶
An unbiased unsupervised type of clustering analysis is a good starting point to familiarize yourself with a new
dataset. The t-SNE algorithm is an algorithm that was developed in recent years. It finds similarity in expression profiles of
samples and will place cells with similar expression profiles together on a map. UMAP is a more recent, faster and often considered better alternative to t-SNE that groups similar cells together while also maintaining an overall sense of how different groups relate to each other. You will find options for both algorithms in R2.
In R2, these maps can be generated by users with an account. Once a dataset t-SNE or UMAP has run, it is available to other users as well. This saves
processing time and costs.
We will use a dataset that was adapted from one of the resources of The Cancer Genome Atlas (TCGA). They provide a wealth of omics data: “TCGA generated over 2.5 petabytes of genomic, epigenomic, transcriptomic, and proteomic data. The data, which has already led to improvements in our ability to diagnose, treat, and prevent cancer, will remain publicly available for anyone in the research community to use.”
- In the left side-menu on the main page, click on Sample maps (UMAP/tSNE)
- In the grid, filter for the dataset Tumor Colon Adenocarcinoma (students) - tcga - 204 - tpm - gencode36 and click its Select button.
Under the graph, a menu allows the user to adapt settings. Colors of the graph points are not set by default.
- Find the Color mode dropdown and select Color by Track. Now set the Color track dropdown to use the cms_predicted (4 cat) track, and click Set colors to show the changes.
- Note that you can exclude track subgroups in the legend. E.g. click on the yellow square from the legend to hide data points with that value. Click the legend title to invert the data point selection. Click the boxes of the hidden track value data points to show them again.
The most important parameter for the t-SNE algorithm is the perplexity value. The perplexity parameter controls the balance between a focus on preserving local details or global structures of the data. When R2 receives the request to calculate the t-SNE map for a dataset, it immediately calculates and stores the t-SNE maps with different perplexity values. The resulting maps can be found under the setting Version. It is also possible to show all the available perplexity maps for the dataset at the same time.
- Set Version to the value all and color again by the same track (or try out the coloring by the expression of a gene if you prefer so).
![]() What insight did you obtain when you colored the plot with annotation?
What insight did you obtain when you colored the plot with annotation?
![]() You looked at several embeddings of the t-SNE plot that were created with different perplexity values.
Roughly, what is your impression of the clustering of the samples. Why do you think it is good practice to check different values for a parameter?
You looked at several embeddings of the t-SNE plot that were created with different perplexity values.
Roughly, what is your impression of the clustering of the samples. Why do you think it is good practice to check different values for a parameter?
Let’s see if these CMS subtypes, that cluster separately on the t-SNE map, hold any prognostic survival value.
1.3.2. Different survival chances for different CMS CRC subtypes?¶
We will use the Guinney dataset: Tumor Colon (CMS) - Guinney - 3232 - custom - ccrcst1
- In the left side-menu on the main page, click on Survival (Kaplan-Meier / Cox)
- In the menu at the center of the page, select a dataset by clicking on Select a data set, and find the dataset of which the amount of samples N is 3232 and Author Guinney
- Click on the row to read its description in the information box underneath the dataset selection grid and confirm
- Leave Separate by at categorical track (Kaplan-Meier) and click Next
- Choose Type of Survival overall and Track cms_final and click Next
- Now perform the same analysis, for this you can click Kaplan start button in the top-left of the screen. This time choose relapse-free instead of overall for the setting type of Survival
![]() What does the first Kaplan Meier plot tell you? (You can still go back to the previous plot by clicking the back-button in your browser)
What does the first Kaplan Meier plot tell you? (You can still go back to the previous plot by clicking the back-button in your browser)
![]() And what is your conclusion from the second Kaplan Meier graph?
And what is your conclusion from the second Kaplan Meier graph?
1.3.3. Mutations¶
Now we would like to look into colorectal cancer associated mutations and see if they are specific for one of the CMS groups
- On the main page, select the Guinney dataset if it is not selected in box 2 yet
- Choose the relate 2 tracks analysis to show the different ratios of mutations per CMS subtype and click Next.
- For the X track choose cms_final (5 cat) and for the Y track choose mutant_braf (3 cat) mutations.
- Select the Graph type Stacked bar (%)
- The Guinney dataset contains several datasets put together. To only look at the samples from studies that looked at the mutational aberrations, use the Sample Filter with the setting Subset Track, select mutant_braf (3 cat) and in the pop-up check the boxes of 0 (776) and 1(87). Note that the value 0 stands for ‘braf mutation: no’ and 1 for ‘braf mutation: yes’ and nd stands for ‘no data/ not defined’. We thus eliminate the samples from this analysis for which it is not known whether they have a BRAF mutation (value nd gets omitted). Click Ok and then Submit.
![]() Which CMS subtype shows the highest frequency of BRAF mutations?
Which CMS subtype shows the highest frequency of BRAF mutations?
Now we will look at the KRAS mutations
- Underneath your stacked bar plot, scroll down to the Adjustable settings menu, change the y-axis to mutant_kras.
- Use the red cross behind the setting Subset Track to eliminate the BRAF mutation subset and click on the dropdown to now select mutant_kras. In the pop-up check the boxes of 0 (560) and 1(336) (thus omit the samples with ND values), click Ok
- Click Submit to see the result
![]() **Which CMS group shows the highest amount of KRAS mutations? What difference do you notice in the **
**Which CMS group shows the highest amount of KRAS mutations? What difference do you notice in the **
1.3.4. A dive into CMS1: MSI / MSS in CRC¶
Colorectal cancer exhibits two primary forms of genomic instability: chromosomal instability (CIN) and microsatellite instability (MSI), with MSI being a hallmark of the CMS I subtype. MSI arises from defects in DNA mismatch repair genes, which are responsible for correcting errors that occur during DNA replication. When genes such as MLH1, MSH2, MSH6, and PMS2 are mutated or silenced, the repair process fails, leading to the accumulation of mutations in short repetitive DNA sequences known as microsatellites. The presence of MSI predicts a good outcome in colorectal cancer.
In MSI colon cancer, genes of the DNA mismatch repair system thus play an important role. Germline mutations in these genes are a major cause of the inherited form of colon cancer, namely HNPCC (hereditary nonpolyposis colon cancer). In sporadic forms of colon cancer however, these genes are frequently inactivated. Inactivation is often achieved via hypermethylation, switching the gene off. Hypermethylation of genes in colon cancer is most common in colon tumors with a proximal location in the colon and much less in colon tumors with a distal location.
The next section we will use another dataset. “Tumor Colon- Watanabe - 84 - MAS5.0 - u133p2”
This dataset consists of Microsatellite Stable (MSS) tumors and Microsatellite Instable (MSI) tumors.
MSS vs MSI¶
- From the main page, find the dataset Tumor Colon - Watanabe - 84 - MAS5.0 - u133p2 and read the Summary in the information panel underneath the dataset selection grid.
- Then Confirm selection of the dataset.
- Use the Differential expression between two groups module to generate a list of differentially expressed genes between MSI and MSS characterized tumors (group by MS_status).
Because we know that DNA repair genes play an important role in microsatellite (in) stability, we can use a set of DNA repair genes to examine whether these genes indeed are differentially expressed between MSI and MSS tumors and which genes exactly make the difference.
- With the Select gene set button, filter for DNA repair genes, and find them in Categories. There are 247 genes annotated as DNA repair genes. Perform the analysis by clicking on Submit.
One of the genes that is differentially expressed, is MLH1.
- Click on its magnifying glass to look at the expression of MLH1 in the MSI vs. MSS samples.
![]() In the list of differentially expressed genes, MLH1 is significantly lower
expressed in the MSI group vs the MSS group: MSI < MSS.
In the list of differentially expressed genes, MLH1 is significantly lower
expressed in the MSI group vs the MSS group: MSI < MSS.
When you look at the expression plot of MLH1, what do you notice about the expression of the samples in this group? Do you see a clear-cut trend of lower MLH1 expression in the
MSI group?
MSI tumors give a very heterogeneous picture. This could be an indication that within the MSI tumor group also a subgroup could be identified.
- Hover with your mouse over data points to see if you can identify a subgroup of an annotation track in which low MLH1 expression occurs more often.
- We can add an extra layer of information by coloring the dots of the violin plot. Scroll down to the Adjustable settings menu and set Color mode to Color by a Track and Color track to MS_Orientation (5cat), and submit.
To find out if our gene of interest associates strongly with any annotation that is already available for the current dataset, R2 also offers a tool: CliniSnitch. CliniSnitch performs a context-dependent statistical test on each track to identify significant associations with the gene’s expression (i.e. different types of tests based on whether a track is numerical or categorical).
- On the right hand side of the page, you find several followup links in light grey boxes with dark grey headers. In one of them you can find CliniSnitch. Click on the CliniSnitch MLH1 link to find out which available track shows the strongest association with MLH1 expression.
- Click on the magnifying glass icon in front of the track with the lowest p value to find out which subgroup it is that we are looking for.
![]() Concluding, with which two characteristics can you describe the subgroup that
more often has a low MLH1 expression?
Concluding, with which two characteristics can you describe the subgroup that
more often has a low MLH1 expression?
In many cases of proximal colon cancer with MSI, the high level of microsatellite instability is caused by the loss of MLH1 expression. Understanding the relationship between MLH1 expression and MSI is crucial for diagnosing MMR (mismatch repair) deficiency, predicting prognosis, and guiding targeted therapies for patients with colorectal cancer.
Colorectal tumors with MSI do not have the same response to chemotherapeutics and are an important subgroup to look at for specialized treatments. In the Watanabe data set we found MLH1 as possibly an important player. We will now corroborate our findings in another dataset. Not only because this is an old set (look up the background information in the information panel of the dataset selection grid), but it is common practice to validate your results with other sources. We will use a dataset that we already looked at with the t-SNE clustering algorithm.
MSI in tcga set¶
- Go back to the main page, and select Tumor Colon Adenocarcinoma (students) - tcga - 204 - tpm - gencode36
- Perform the Differential Expression between two groups analysis, choose Track microsatellite_instability, click next, set no for Group1 and yes for Group2, and perform the analysis on the gene set Broad 2025 06 c6 oncogenic. Submit
- Click again on the MLH1 gene magnifying glass.
In this set too MLH1 seems to play a key role for MSI related CRCs. It would be interesting to find out which genes are possibly regulated by the MLH1 gene. One way to find genes that are possibly affected by MLH1 is to look at (inversely) correlated genes.
Find genes correlating with a single gene¶
- From the main page, run the Find Correlated Genes with a single Gene module for the MLH1 gene. Use again the filter option for the Broad 2025 06 c6 oncogenic gene set.
- On the result page, click on the best correlating gene (not MLH1) to plot both genes together, in a two gene view. Inspect the correlation. Can you think of reasons why the gene expression might be highly correlated?
- Click on View Additional Details, look at the Probeset Genome Location table.
On which chromosomes are both genes located?
- In the table, click on one of the two T-view links and zoom out 2X with the correct button at the top of the page.
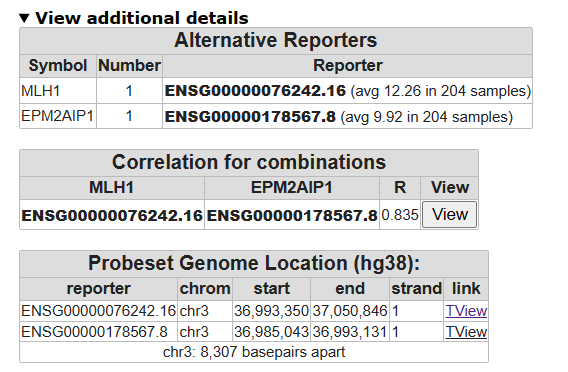
You arrived at the Genome browser. The Genome Browser allows you to “walk over the genome”.
Underneath, each genome location is annotated with the genes found at that position. Genes colored in red are read in reverse direction.
![]() What can you say about the location of the two genes?
What can you say about the location of the two genes?
Now let’s see how strongly MLH1 expression is associated with CMS subtypes.
- From the main page, select the Correlate Gene with a track analysis to confirm the strong association of MLH1 expression with the CMS1 subtype (track cms_predicted (4 cat)).
DNA methylation¶
Inactivation of a gene can occur due to mutations in the gene or through epigenetic changes, such as promoter methylation. The R2 platform hosts a variety of dataset types. Not only gene expression datasets but also methylation arrays can be found.
- Go to the main page and select the dataset Tumor Colon adenocarcinoma - tcga - 296 - custom - ilmnhm450
- Create the View a Gene plot for MLH1
- Under the triangle of View additional details click on the view all link.
- Scroll down to the bottom of the page to make a Subset Track selection with microsatellite_instability to only select the samples that have been annotated for this characteristic (yes and no). Click Next.
A heatmap is generated of methylation levels. Each column corresponds to a sample, and each row corresponds to a feature (a single CpG site or a larger target region including multiple CpG sites, e.g. promoter regions). A hierarchical clustering of the samples is performed. The MLH1 reporters themselves seem faulty (complete blue colored rows), but you find CpG sites above that are located at the promotor sites of MLH1 and the gene that we have seen before as strongly correlating in expression. Note, that blue is methylated and yellow is non-methylated.
- Hover with your mouse over the r2_at_cms track to see to which CMS subtype the cluster of hypermethylated samples mostly belong.
![]() Which CMS group did you find?
Which CMS group did you find?
![]() Why are we looking at methylation levels around the promotor of MLH1?
Why are we looking at methylation levels around the promotor of MLH1?
1.3.5. What pathways drive subtype CMS4?¶
Previously we looked into CMS subtype 1. Let’s now have a look at the other subtypes. We would like to understand what sets CMS 4 apart from the subtypes 2 and 3.
- On the main page, select Tumor Colon Adenocarcinoma (students) - tcga - 204 - tpm - gencode36
- Choose the analysis Differential Expression Between Two Groups.
- Choose the track cms_predicted, click next and look at groups cms2 and cms4
- The analysis can take a while.
Gene set analysis helps researchers interpret the biological relevance of a group of genes. Instead of looking at individual genes, it allows you to understand the collective functions or pathway involvements of genes in your list. This can provide more meaningful insights into the underlying biology of a particular condition or experiment.
- Click on the topmost button on the right that is labeled Gene set analysis.
- Select the Gene set Collection Custom collection and select Gene set Broad 2025 06 h hallmark
- Switch the Representation setting to all to look at both over- and under-representation
- Click Next
![]() Which gene sets do you recognize from the first column of the table? Name a few that are overexpressed in CMS 2 and name a few that are overexpressed in CMS 4. On top of the table you find the meaning of the color coding green and red
Which gene sets do you recognize from the first column of the table? Name a few that are overexpressed in CMS 2 and name a few that are overexpressed in CMS 4. On top of the table you find the meaning of the color coding green and red
![]() Explain the biological relevance for the CMS4 subtype for these over- or/and underexpression of these gene sets for CMS4 subtype CRC tumors
Explain the biological relevance for the CMS4 subtype for these over- or/and underexpression of these gene sets for CMS4 subtype CRC tumors
1.4. A song of Genomic Alterations en Oncogenes¶
Thus far we looked at colon cancer characteristics mainly based on gene expression. Recently, a new article was published that describes and analyses a large cohort of CRC patients. The data used in this new article include both genome and transcriptome information. Notably, this dataset is exceptionally well-annotated with extensive clinical information (Nature 2024: CRC : Nunes et al). It contains 1063 tumor samples and 120 tissue normals.
1.4.1. Introduction to the Nunes data¶
It is good common scientific practice to validate and check your analysis in other resources. We already have seen that the Molecular signature classification (CMS) are nicely clustered in the previous datasets. Let’s check whether this is also the case for the Nunes RNA seq dataset.
- Select on the mainpage Sample Maps (UMAP/tSNE).
- Find and click on the UMAP embedding of Tumor Colon - Nunes - 1063 - tpm - ensh38e98 to read the Summary of the dataset in the information panel under the grid and then Select the dataset.
- Use Color mode again to overlay the plot with Color by a Track and cms_tumour (5cat).
- If you want, you can check multiple (or all) versions
Again the CMS clusters clearly separate this set into 4 clusters, confirming our previous conclusion that CMS subtypes have unique gene expression patterns. This indicates that they might refer to biologically different subtypes.
- Going back to the main page of R2, make sure that the Nunes 1063 dataset is selected in box 2.
- Also check in the Cohort Overview the annotation to glance over the kind of annotation that is provided. Think for yourself what kind of questions you could explore with the annotated data at hand.
![]() In context of using the genomic data that belongs to this dataset, which annotation would yóu be interested in to further examine and why? Just name a few annotation tracks and provide a short explanation what you want to look investigate.
In context of using the genomic data that belongs to this dataset, which annotation would yóu be interested in to further examine and why? Just name a few annotation tracks and provide a short explanation what you want to look investigate.
Genomic data¶
Now let’s dive into the genomic data.
In the context of colorectal cancer, Comparative Genomic Hybridization (CGH) profiles are used to detect copy number variations (CNV) across the genome. Those CNVs include amplifications (gains) and deletions (losses). This type of genomic events can lead to overexpression of oncogenes and or loss of tumor suppressor genes.
 Example of CGH profiles: CGH profile of malignant melonama. A is a control sample
Example of CGH profiles: CGH profile of malignant melonama. A is a control sample
CGH profiling plays a crucial role in understanding colorectal cancer (CRC) by revealing genomic instability, a hallmark of cancer. We uploaded the genomic data of the Nunes study into the platform, and can generate CGH profiles of the .
Let’s examine these profiles to see if any patterns emerge when plotting them according to different characteristics.

- On the main page select in the left menu WGS/WES data and in the middle menu click on Plot CGH karyograms in track assisted order. Select CRC Nunes 2024 (copy number) and click Continue.
An averaged CGH plot, integrating the CGH data from all 1063 tumor samples, provides a summary of the gains (in red) and losses (in blue) observed across the dataset in a single visualization.
Now, let’s examine the CGH profiles for each CMS subtype.
- In the second pull down X track, select cms_tumour (5 cat) and click Redraw.
![]() When examining the CGH profiles for the different CMS subtypes, what patterns or variations do you observe?
When examining the CGH profiles for the different CMS subtypes, what patterns or variations do you observe?
In cancer, a high rate of mutations can occur in the genome and could sometimes even reach tens or hundreds of mutations per megabase. In general, tumors are classified as hypermutated when they exhibit more than 10-12 mutations per megabase.
This dataset is annotated for hypermutation status.
- Generate CHG profiles split by hypermutation status: change X track to hypermutation_status (2cat). Don’t forget to Redraw. You can also check the msi_status (2 cat) track.
![]() What do you observe? Can you think of an explanation for the differences that you observe?
What do you observe? Can you think of an explanation for the differences that you observe?
- Go to Relate 2 tracks in te main menu and select the hypermutation status track for one of the axes and the track of the CMS classification for the other. Choose a Stacked bar % Graph type and hover with your mouse over the bars to read the respective percentages.
Don’t be shy to play around with any other annotations to gain more insight in the characteristics of subtypes in this dataset (e.g. MSI vs CMS ). Be aware that you check these conditions in a specific dataset, and pause to think if this corresponds with knowledge that you gained from literature or lectures.
CMS2 subtype has a significant lower incidence of tumors that were classified as hypermutated. CMS1 has the highest amount of samples that were hypermutated. We can utilize R2 to explore the specific mutations present, along with detailed information on their types—such as missense, nonsense, frameshifts, and more.
- Go the main page, click in the left menu on WGS/WES data and select Pers. Med. OncoPrint (fixed data) and as resource the CRC OncoKB hotspots Nunes.

In this OncoPrint, we find an overview of the most common mutations across our entire cohort. Samples are shown in the columns, the genes are shown on the rows. The most commonly mutated genes are found on top. The color coding can be found underneath the plot and is seen in the image below as well. Thus, the colors in the plot represent the type of mutation per sample per gene. Above the OncoPrint, you see one bar of annotation, which is the CMS annotation.

OncoPrint color legend: the colors represent the type of mutation
Scrolling down to the bottom, you will find in the Adjustable Settings menu all sort of interesting options that you can toggle on/off. In the picture below, you can find several suggestions to reveal more insights of mutational characteristics in this cohort and of CRC in general. You can add extra tracks to the plot if you click on Select Tracks.

![]() Name the four top genes. Also check with the CMS subtype annotation above the plot if you can relate the information in the plot with your knowledge about CMS subtype specific mutations.
Name the four top genes. Also check with the CMS subtype annotation above the plot if you can relate the information in the plot with your knowledge about CMS subtype specific mutations.
Discuss with each other how mutation of the four top genes plays a role in CRC. Check which mutation types are often found in the four top genes.
1.4.2. Somatic variants¶
Let’s investigate mutations in more detail and look at individual sample mutations in this cohort. On the main page in the left-side menu, select the WES/WES data module and then select Somatic Mutations.
- Select as variant hg38 - CRC nunes (Nature 2024) OncoKB only. Click Next.
Immediately you see a list of genes that should look familiar.
- Now switch to detailed in the Detailed or summary setting and click Next.
In the grid you can find all the reported mutations for genes that are part of the OncoKB database (https://www.oncokb.org/about). The details of each mutation can be found in the columns.
If you click on the view (!! not detail, you might have to broaden the first column’s width) links in the first column of a mutation, this will guide you to the genomic location of the mutated gene in the Gene browser or the Genome Browser respectively.

Click the view link! You might have to drag the Link column wider
When you clicked on the View link of the first mutation to go to the GenomeBrowser at the APC location, you can zoom out 20x twice with the navigation button on top. You will then see all the different reported mutations for the APC gene. By hovering over the small colored blocks, you can examine the mutation class and view details about the specific nucleotide changes and their effects.
KRAS and BRAF mutations are oncogenic driver genes in cancer.
- Use the top left Find Gene box to find KRAS. Choose the top KRAS result to jump to its genomic location and to look at the somatic variants.
- Also look at BRAF.
![]() If you look at the complete gene locations of APC vs BRAF / KRAS and the somatic variants in the GenomeBrowser, do you see any difference with respect to how many variations are found in the cohort?
If you look at the complete gene locations of APC vs BRAF / KRAS and the somatic variants in the GenomeBrowser, do you see any difference with respect to how many variations are found in the cohort?
Which mutation type in BRAF mutation is the most prominent?
So far, we have examined the transcriptome and genomics data separately, with the only overlap being some annotation of hypermutation status derived from the genomics analysis.
For your information: in R2 you can easily use the somatic variation module to annotate each sample with the mutation status in a cohort: you can use the filters in the header of the Gene column to filter a specific gene and then use the button Build track underneath the grid to annotate each sample with its mutation status for that gene.
This way you can annotate your dataset for the presence of any gene mutation / specific type and use this annotation with gene expression dataset(s).
Below is pictured how you can use these tracks (annotations) to find differentially expressed genes, similar to how you have already performed such analyses in the previous exercises. For now we continue to another exercise.

1.4.3. Signature scores¶
Even though it can be very interesting to look at the transcriptome effects of a specific gene mutation, you might want to look at several genes at once. In R2 we do this with signature scores.
Within the current context, we define a signature as a collection of genes that are defined on a particular basis. This can be the presence within a gene-ontology class, the genomic location of a gene, or perhaps something potentially more meaningful like a functional pathway signature.
To summarize the expression values of the genes in such a signature, R2 can calculate a signature score. This signature score is simply defined as the average z-score of a z-score transformed dataset (the standard way of visualizing a heatmap). In R2, such scores are automatically generated when one generates heatmaps via the “View a geneset” function.
Often you can find multiple signature scores:
- Average expression of the signature genes.
- Weighted sums where each gene’s contribution is weighted by its known importance.
A Differential Expression between two groups analysis has already been performed for APC mutants versus non-APC mutants for the Nunes set. We have stored a list of genes which are upregulated in the APC mutated group. This collection of genes will be used to find correlations for the signature scores of other pathways.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- Go the main page and select the Geneset vs Genesets Correlations module. In the next screen select for the Gene set (source) a sub gene set: click on the arrows in front of the geneset collection to go deeper in the tree Community genesets > r2 > main > APC_activated (153) (click the green confirm button).
- For Gene set (target) select the complete collection of Broad 2025 06 h hallmark genesets.
R2 will calculate the signature scores for the upregulated APC mutant gene list and for each gene set that the selected Broad Hallmark gene set collection harbors.
![]() Which pathway do you expect to pop-up as a result of this test?
Which pathway do you expect to pop-up as a result of this test?
Click next. R2 is now performing a correlation test on the signature scores of the APC activated gene list in APC mutants against the signature scores of all the gene sets in the Broad hallmark collection.
![]() Name other pathways in the list that you recognize and that can be associated with CRC or CMS classes.
Name other pathways in the list that you recognize and that can be associated with CRC or CMS classes.
![]() Several expected CRC-related pathways appear in the table, suggesting potential pathway involvement, though this is only indicative and not necessarily causal. What experimental approaches would you propose to investigate the effects of a specific mutation?
Several expected CRC-related pathways appear in the table, suggesting potential pathway involvement, though this is only indicative and not necessarily causal. What experimental approaches would you propose to investigate the effects of a specific mutation?
1.5. Therapeutic: Effects of imatinib: shifts of signature profiles and molecular subtypes¶
Mesenchymal Consensus Molecular Subtype 4 (CMS4) colon cancer is associated with poor prognosis and therapy resistance. In a proof-of-concept study, Kranenburg et al. assessed whether the drug imatinib could shift the CMS4 subtype specific gene expression program towards a gene expression program that is better treatable.
The button below brings you to the form in which you can submit your answers for the third section.
We want to see how the gene expression of specific mesenchymal genes, such as ZEB1, PDGFRA, PDGFRB, and CD36, changes between pre- and post-imatinib treatment samples:
- On the main page in the center menu, select the dataset Tumor ImPACCT - Kranenburg - 30 - custom - ensh37e75
- Choose the analysis View Multiple Genes and click Next
- In the Genes/Reporters to include textbox, type Zeb1,PDGFRA,PDGFRB,CD36
- Set Track to imatinib (2 cat) that divides the samples in a pre-treatment and a post-treatment group
- Click on the blue gear icon in the upper left corner of the plot and change Handle groups by Separated by track grouped by gene.
- Set Color mode (groups) to Color by Track in order to make the box plots visually more distinct.
- Check Add Scatter to see the data points
- Note that you now also can change Color mode of the scatter points to color by track or by gene (e.g. CTNNB1)
- The plot options in the panel that pops up with the gear icon are directly responsive. Therefor, you don’t have to submit your changes for these options, which ís required when you make adaptations in a adjustable settings menu. Sometimes when you change the fonts too much, the plot might look a bit out of balance. Then you can still click redraw plot that often rearranges plot elements for the better.
![]() What can you say about the level of expression of these genes post treatment?
What can you say about the level of expression of these genes post treatment?
![]() What is the role of ZEB1 in EMT? (Use any informative source on the internet)
What is the role of ZEB1 in EMT? (Use any informative source on the internet)
What is the biological implication of this experiment?
1.5.1. Proliferation vs metastases¶
Mesenchymal tumor phenotypes are generally accompanied by reduced proliferation. Indeed, high expression of proliferation signatures and Wnt target genes are associated with good prognosis and reduced metastatic capacity in CRC
- On the main page, make sure that the selected dataset is Tumor ImPACCT - Kranenburg - 30 - custom - ensh37e75
- Select the analysis View Geneset (Heatmap) and click Next
- In the Gene set selection grid, search for HALLMARK_MYC_TARGETS_V1 (200) and select it by clicking on the triangles in the Broad 2023 10 h hallmark collection until you’ve found it. Check the gene set and Confirm selection
- Submit
The heatmap for the z-scores of the expression values of the MYC targets geneset is shown. Underneath the heatmap you find colored squares which denote the geneset average z-value per sample, the signature score. With an account you can save such scores as a Track to use in further analyses in R2.
- Click on the Store (R2) link in the small table, a bit underneath the heatmap.
- In the page that pop-ups, you can adjust settings
- Check the name that is provided for this signature score: hallmark_myc_targets_v1 and read the description
- Note that you can change teh setting Where from Temporary (24h) to personal track, in case you want to store it long term. Or you could choose to store it to a community if you are part of one and want to share this Track with your colleagues. We leave everything as is and click on Build set
Now that we have an average expression value for the MYC target genes per sample, we would like to assess how the treatment influenced these values. - Go back to the main page
- Click Relate 2 tracks, click Next
- Select X track imatinib (2cat) and for the Y axis we choose the signature score that we just generated: Y track hallmark_myc_targets_v1
- Select Graph type Dot
- Change Color mode (groups) to Color by Track and click Submit
We also would like to know the influence of the treatment on WNT target gene expressions. - Do the same route, starting with View Geneset (heatmap), for the WNT target gene set: find WNT_ImPACCT that you can find in the gene set collection Community gene sets (use the search field of the grid) and store the signature score as a track as well.
- Again perform the Relate 2 tracks just as in the previous exercise, this time with your WNT target gene signature on the Y axis.
- Optionally change the Graph type to another type that you want to try out
![]() What happened with the expression of WNT- and MYC-target genes post treatment?
What happened with the expression of WNT- and MYC-target genes post treatment?
![]() Even though it is counterintuitive, can you think of a reason why this actually
could be a better outcome than before treatment?
Even though it is counterintuitive, can you think of a reason why this actually
could be a better outcome than before treatment?
1.5.2. Assess the prognostic value of imatinib treatment¶
To assess the potential prognostic value of the treatment, we will make a signature of the genes that were changed after treatment.
- On the main page, make sure that the selected dataset is Tumor ImPACCT - Kranenburg - 30 - custom - ensh37e75
- Select the analysis Differential expression between two groups
- Switch the Group by setting to imatinib (2cat) and click Next
- Choose Group 1 pre-imatinib (15) and Group 2 post-imatinib (15)
- Set the P-value cutoff to a stricter value: 0.001 and click Submit
A table shows the differentially expressed genes. On the right underneath buttons with followup analyses, you can find a small table that shows how many genes were downregulated by the imatinib treatment (imatinib: pre-imatinib >= post-imatinib) and how many genes were upregulated (imatinib: pre-imatinib < post-imatinib).
![]() How many upregulated genes were found?
How many upregulated genes were found?
- To use this gene list in other analyses within R2, click on the lowest button on the right side that is labeled Store result as custom gene set

{kind=link}
- As a name, type impacct_imatinib_treatment_up
- In the Included groups check only the upregulated genes
- Click on Save gene set
The treatment resulted in a shift in gene expressions. To find out what the effect is of this shift, we will make use of the geneset of upregulated genes that we just saved, now in combination with the Guinney dataset, the cohort dataset with annotated CMS status and survival data. We use the unsupervised k-means algorithm to find groups in our cohort that show similar expression patterns for our geneset.
- On the main page, select the Guinney dataset with 3232 samples again
- Select the K-means analysis in box 3 and click Next
- In the Subset Track dropdown, select stage, and in the pop-up window check the boxes 2 and 3, click Ok
- Behind the setting Gene set, click Select gene set to find your previously stored gene set under Personal gene sets > _TEMPORARY > impacct_imatinib_treatment_up. Check the box in front of your gene set and Confirm selection.
- We leave the number of groups at 2
- Set the Cell width to 1 and Cell height to 8 and click on next
The Kmeans algorithm looks at the expression of the samples for the selected genes and makes two groups of samples that show most similar expression patterns. Then for each gene it shows the expression by a color code
![]() Which color group would you say shows high expression and which color group shows
low expression of the geneset?
Which color group would you say shows high expression and which color group shows
low expression of the geneset?
(Thus, high: yellow or purple and low: yellow or purple)
Again this group division can be stored in R2 to use in a next analysis.
- To do so, hit the button Store as track that you can find on the left above the k-means heatmap
- On the following page, just click the button Next
- To save the results in a way in which we will easily remember what the track was for and which group showed which expression, change the name of Group ‘cluster 1’ into high and of Group ‘cluster 2’ into low. Also change Track name into kmeans_imatinib_induced
- Click on Create Track and go back to the main page
Let’s see which CMS subtypes are represented in the two k-means sample clusters
- On the main page, select the analysis Relate 2 tracks
- For the X track scroll all the way down and select kmeans_imatinib_induced (3 cat)
- For the Y track choose cms_final (5 cat)
- In the Subset Track dropdown, select stage, and in the pop-up window check the boxes 2 and 3 , click Ok
- Change the Graph type into Stacked bar (%) and use *Color mode (groups) Color by Track
- With a click on the gear icon you can change plot options, such as Order Groups by to group size. Also note the different tabs in the plot options.
![]() If the impact of imatinib shows a shift of the geneset from low expression to
high expression values, what shift in CMS subtypes do we expect to see?
If the impact of imatinib shows a shift of the geneset from low expression to
high expression values, what shift in CMS subtypes do we expect to see?
![]() What is known about the treatability of subtype 4 and cms 2 respectively?
What is known about the treatability of subtype 4 and cms 2 respectively?
- From the main page in the left menu click the Survival (Kaplan Meier/Cox) analysis
- Check that the Guinney set is selected and that the separation is made by a categorical track. Click Next
- Choose overall survival type and Track kmeans_stage23_imatinib_induced (scroll all the way down). Click Next
- In the left menu click again the Survival (Kaplan Meier/Cox) analysis
- Repeat the process but select the relapse free instead of overall survival type.
![]() What is your conclusion?
What is your conclusion?
1.6. (OPTIONAL) Identifying key drivers of CRC: superenhancers controlling gene expression¶
An enhancer is a 50-1500 bp region of DNA where activator proteins, such as transcription factors, bind and that
increase the likelihood transcription will occur at a gene. They can be located up to 1 Mbp away from the gene,
either upstream or downstream from the start site, and either in the forward or backward direction. A super-enhancer
consists of multiple enhancer regions. This larger region is bound by more transcription factor proteins to drive
transcription of genes. They can be up to 20 times the size of an enhancer.
In chapter Integrative analysis: ChIP-seq data of the R2 Tutorial, you can find a more detailed description of Chipseq data analysis.
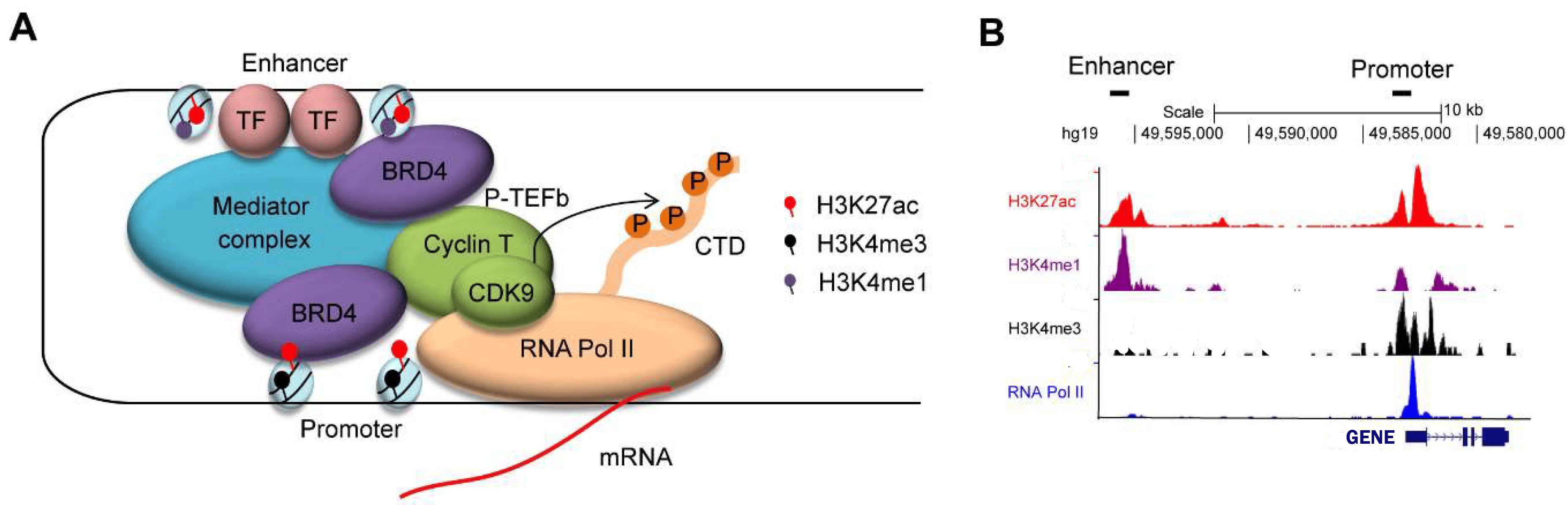
The histone modifications of active promoter sites and (super-)enhancers of a gene
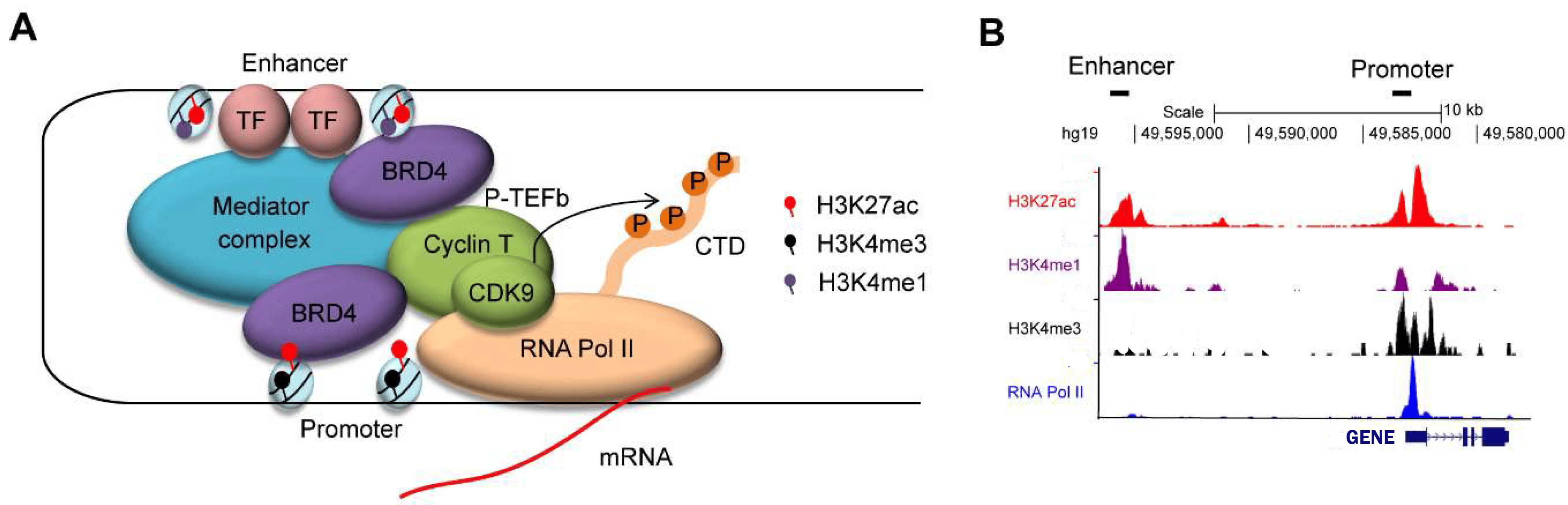{kind=link}
Enhanced enhancer activity can lead to the overexpression of oncogenes, which promotes cancer growth. Super-enhancers often play a central role in determining cell identity and tumor initiation and progression. Identifying these active enhancers can help pinpoint key drivers of colorectal cancer, potentially revealing new therapeutic targets.Different patients may have colorectal tumors with distinct enhancer landscapes. By characterizing enhancer activity, researchers can potentially classify patients into subgroups with different treatment responses or prognosis, enabling personalized medicine approaches.
With Chromatine Immuno Precipitation binding of elements to the genome can be studied. Transcription of DNA to RNA
is regulated by the binding of these elements. These can be Transcription Factors, that bind temporarily to start
transcription, but also chemical modification of the histones (molecular structures that coil the DNA) by methylation, acetylation, etc. These modifications change the accessibility of the DNA for transcription.
When a specific antibody is used in ChIP-seq that recognizes these chemically modified regions, these specific
regions can be studied. Regions with H3K27Ac acetylation mark active enhancers and active promotors (i.e. active
transcription), H3K4Me3 methylation marks active promotors. Studying the relative contributions of both types of
modifications allows a researcher to discern enhancer regions from active transcription sites.
- From the main page left side-menu, select the module ChIP data and click on ChIP Genome Browser
You are now at the Genome Browser at the genomic location of the gene MYCN. Regions encoding genes are drawn at the bottom of the graph. When in red they are encoded in the reverse direction, coding exons are darker.
Li et al. (2021) sequenced 73 pairs of colorectal cancer tissues and generated 147 H3K27ac ChIP-Seq, 144 RNA-Seq, 147 whole genome sequencing and 86 H3K4me3 ChIP-Seq samples. The patients were classified into the 4 CMS subtypes. Therefore, we now have gene expression and chipseq data of this CMS classified patient cohort. Because it is difficult to look at many profiles at the same time, we averaged the data per CMS group. This way we created so called ChIP seq meta profiles. Let’s load them into your Genome Browser
- In the right upper corner, click the button Load / Store Profile
- In the Profile dropdown, select student - Li_CRC_normal_tumor_cms1234 and click Execute.
- Click on the button Goto the GenomeBrowser at the top.
You now see the meta profiles of normal colon tissue, and CMS 1 to 4 meta profiles. For each group you see both H3K27ac and H3K4me3. If you scroll down, you can see that you are at the location of the MYC gene. At an active promotor site, you will find H3K4me3 peaks. You will also find peaks in the H3K27ac profiles at that location. But at an active enhancer site, you will only find H3K27ac, no H3K4me3. Thus left from the myc gene location, you see H3K27ac peaks without H3K4me3, and that is a known superenhancer location of MYC. Let’s look for more superenhancers:
- In the left upper corner, you can write the name of a gene, and you will be taken to its location on the genome. Type ascl2 in the textfield and click Go
- Click the View button (if there are more View buttons, just take the top one) and scroll down to check that you have arrived at ASCL2.
- Now we need to zoom out to find possible superenhancers. Zoom out 10x with the button on top of the page. Then zoom out 5x. Do you see an area with high peaks of H3K27ac where no H3K4me3 can be found? There is a superenhancer.
The difficulty is that you never know how far away from the gene the superenhancer is located. Also, it can be upstream or downstream from the transcription start site of the gene. And… you might even find enhancer regions in the introns of some oncogenes.
Lastly, above the ChIP-seq profiles, you can also find the averaged z-score of the gene expression of the subgroups
of patients. Hover your mouse over the colored dots to see to which group they belong and of which gene it shows the
average expression. This way you can see whether the expression itself differs between the CMS groups, and between
normal colon tissue and tumor tissue.
Ideally you should find oncogenes or transcription factors that are differently expressed in tumor tissue and normal
tissue, and also show different enhancer profiles between normal and tumor tissue.
![]() Play around with the GenomeBrowser. Look for genes that you might remember from
the lectures and see if you can find superenhancer areas. Also check if you can see differences between the CMS
groups, and try to explain the differences. Did you find an interesting gene and how much did you need to zoom out?
Play around with the GenomeBrowser. Look for genes that you might remember from
the lectures and see if you can find superenhancer areas. Also check if you can see differences between the CMS
groups, and try to explain the differences. Did you find an interesting gene and how much did you need to zoom out?
- If you need some inspiration, you could check out genes from this list: IL20RA, LIF, IER3, PLAGL2, FAM3D, TNS1…
- Search the internet as to why these could be interesting genes to look up.
1.7. Evaluation¶
Please fill in the evaluation form about this R2 course!
1.8. Final remarks / future directions¶
This ends the course. Feel free to further explore the course materials or our tutorials.
We hope that this course has been helpful. If you want to visualize and analyze your omics data in the R2
platform, you can always consult r2-support@amc.nl
The R2 support team.